No products in the cart.

Rett Syndrome (RTT) is a rare genetic neurological disorder that predominantly affects females. The condition was first described by Austrian paediatrician Andreas Rett in 1966; the genetic cause was identified in 1999 when Dr Huda Zoghbi and colleagues linked most cases to mutations in the MECP2 gene on the X chromosome.
Rett Syndrome is a neurodevelopmental condition rather than a primarily degenerative disease. Neurones are present but fail to mature and form normal connections, so the disorder reflects atypical brain development rather than progressive neuronal loss. Animal studies have shown partial reversal of some features when functional MECP2 expression is restored, supporting the possibility of targeted therapeutic approaches.
Key Point: In the majority of cases Rett Syndrome arises from spontaneous (de novo) mutations. Most affected children do not inherit the mutation from a parent. Parents are not responsible for the mutation and could not have prevented it.
The condition affects multiple domains: motor skills, communication, cognition and autonomic nervous system function. Common clinical features include loss of purposeful hand use, development of repetitive hand stereotypies such as hand‑wringing or hand‑to‑mouth movements, difficulties with walking, feeding and breathing irregularities, and impaired spoken language.
Severity and progression vary widely between individuals. The clinical picture is influenced by the specific MECP2 mutation and by X‑inactivation in females — a process where one X chromosome is randomly inactivated in each cell, producing a mosaic pattern of cells expressing either the normal or the mutated gene. This mosaicism helps explain why two girls with the same mutation can have markedly different abilities and symptoms.
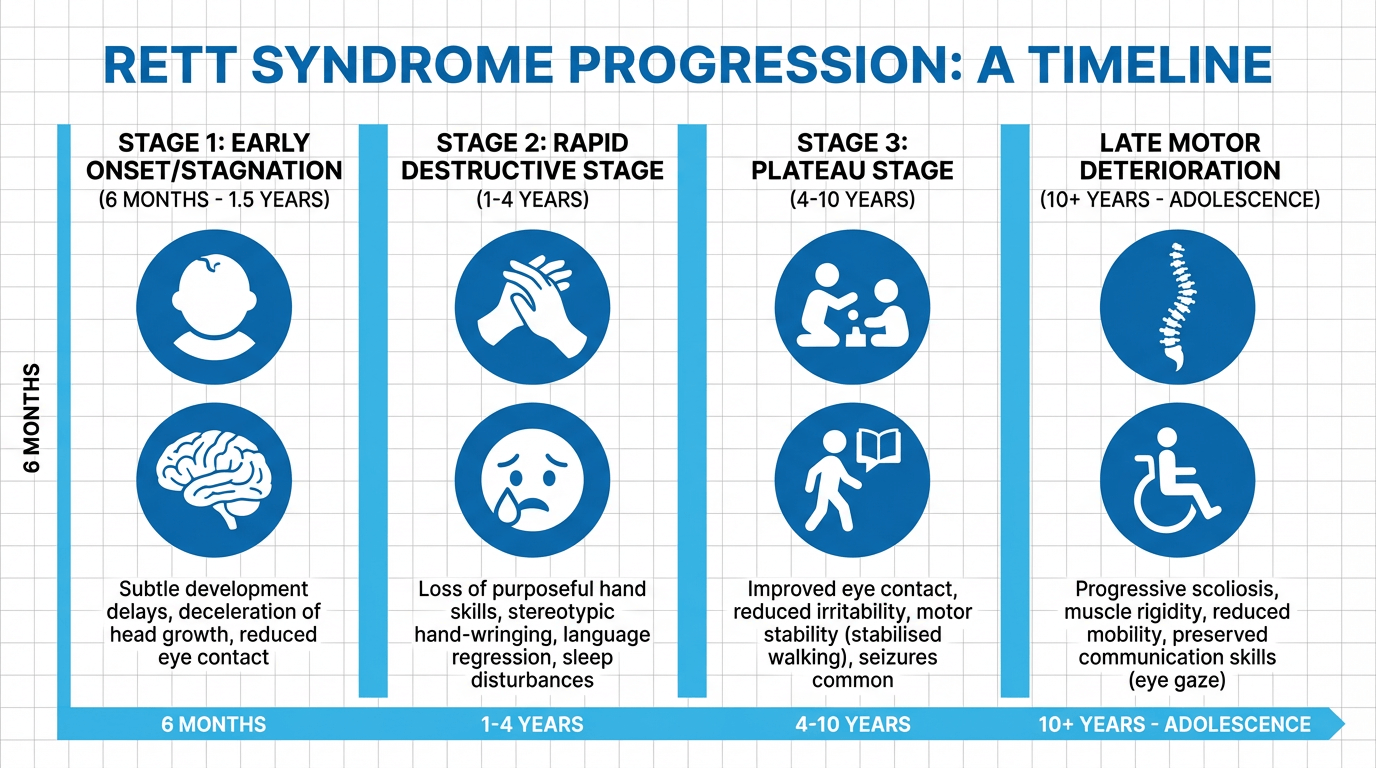 Rett Syndrome commonly follows a recognisable, stage-based clinical course, although not every individual will progress identically. Awareness of these stages helps families and clinicians anticipate changing needs and arrange appropriate interventions, including referral to paediatric neurology if developmental regression is observed.
Rett Syndrome commonly follows a recognisable, stage-based clinical course, although not every individual will progress identically. Awareness of these stages helps families and clinicians anticipate changing needs and arrange appropriate interventions, including referral to paediatric neurology if developmental regression is observed.
Early signs are often subtle. Infants may appear to develop normally for the first six to eighteen months but show reduced engagement and slowed skill acquisition. Typical early indicators include:
Symptoms at this stage are often subtle and may not prompt immediate diagnosis; close developmental surveillance is recommended.
This stage is characterised by rapid developmental regression. Previously acquired purposeful hand use and spoken language are commonly lost. Regression may be sudden or gradual. Key features include:
Breathing irregularities are usually most noticeable during wakefulness and often lessen in sleep. Hand stereotypies persist during waking hours.
After regression, many individuals enter a period of relative stability that can last for years. Some families observe improvements in behaviour and attention, though motor difficulties commonly persist. Features of this stage include:
Stability in cognitive and communicative function is common, although physical support needs often remain substantial.
From late childhood onward, some individuals experience progressive motor decline. This stage is marked by:
Clinical Note: Not all individuals progress through every stage; variant forms of Rett Syndrome may follow different courses. Individual prognosis depends on mutation type and other biological factors.
Alongside the core stages, many individuals develop secondary complications that require active management.
Rett Syndrome is not typically associated with a distinct facial dysmorphism; facial appearance generally resembles parental features. Microcephaly due to slowed head growth may be present. Dental wear related to bruxism and particular gait or posture patterns may be observed.
If regression, loss of skills or any of the signs above are observed, seek referral to paediatric neurology or clinical genetics for assessment and timely management.
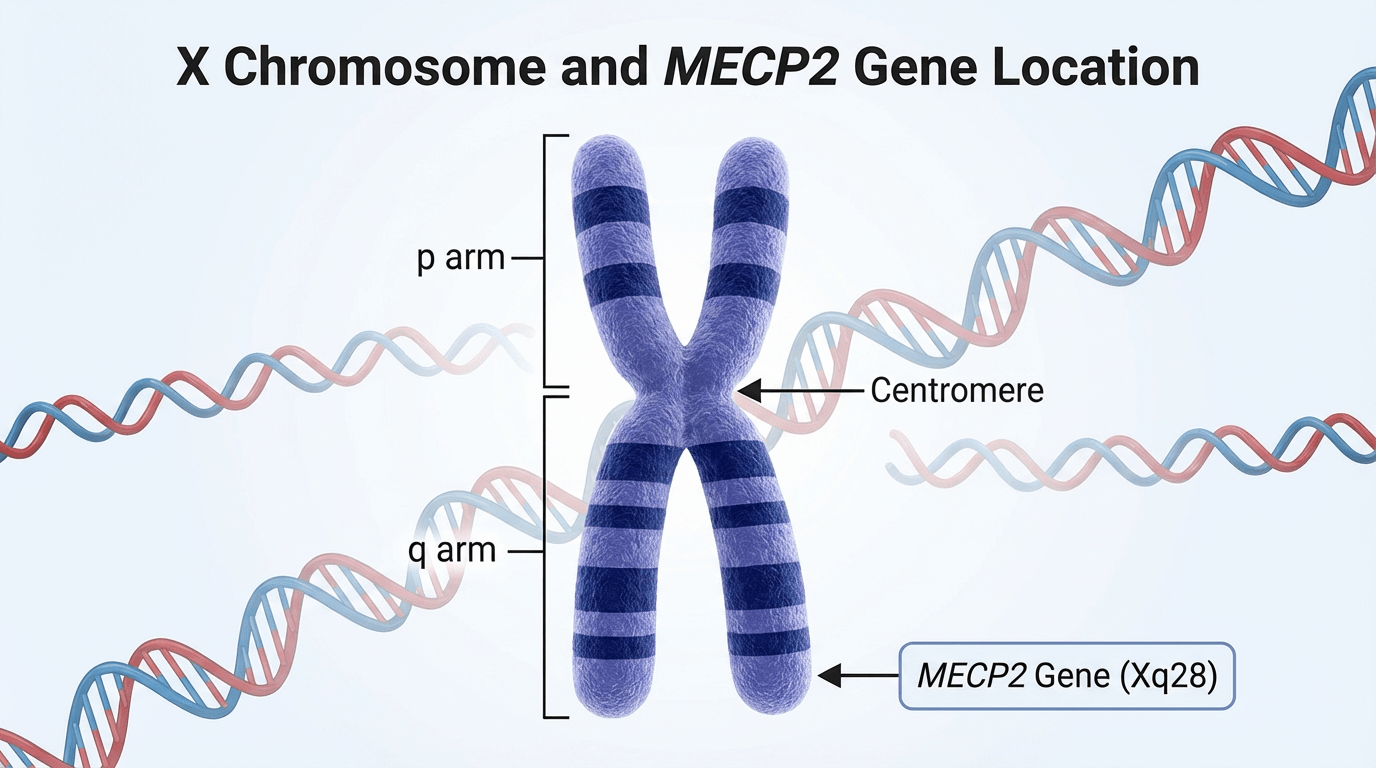 The underlying cause of Rett Syndrome is genetic. The majority of classic cases — approximately 90–95% — are associated with mutations in the MECP2 gene, located at Xq28 on the X chromosome. More than 200 different MECP2 mutations have been described; different mutations may produce variable clinical effects.
The underlying cause of Rett Syndrome is genetic. The majority of classic cases — approximately 90–95% — are associated with mutations in the MECP2 gene, located at Xq28 on the X chromosome. More than 200 different MECP2 mutations have been described; different mutations may produce variable clinical effects.
MECP2 encodes Methyl‑CpG‑binding Protein 2 (MeCP2), a protein that plays a crucial role in brain development and neuronal function. MeCP2 binds to methylated regions of DNA and helps regulate the expression of many other genes, contributing to chromatin organisation, mRNA processing and synaptic plasticity.
MeCP2 is particularly abundant in mature neurones. When MECP2 is mutated, the resulting protein may be absent, reduced or functionally impaired. This alters the normal balance of gene activation and silencing, disrupting post‑natal maturation of neural circuits and leading to the clinical features of Rett Syndrome.
Rett Syndrome follows an X‑linked dominant pattern. Females have two X chromosomes while males have one X and one Y chromosome; this difference largely explains the sex distribution of the condition.
In females who carry a MECP2 mutation on one X chromosome, the other X typically carries a normal copy. Early in embryonic development, a process called X‑inactivation (lyonisation) randomly silences one X chromosome in each cell, producing a mosaic pattern of cells that either express the mutated or the normal MECP2 gene. The proportion of cells expressing the normal gene influences clinical severity.
Males, with a single X chromosome, generally lack this mosaicism. A pathogenic MECP2 mutation on the sole X chromosome usually results in more severe disease; many affected male embryos do not survive to term. Those who do survive often have protective factors such as Klinefelter syndrome (47,XXY), somatic mosaicism, or particularly mild mutations.
Surviving males with pathogenic MECP2 variants may present with severe early‑onset encephalopathy or other MECP2‑related disorders distinct from classic female Rett Syndrome.
Most MECP2 mutations arise de novo (spontaneously) in the parental germline. Parents of an affected child commonly do not carry the mutation and are not at fault. Parent‑of‑origin studies indicate a substantial proportion of de novo mutations originate in paternal gametes, although exact proportions vary between studies.
The recurrence risk for future pregnancies is low for most families but is not zero. Germline mosaicism, where some reproductive cells in an unaffected parent carry the mutation while other cells do not, can produce recurrence; estimates of residual recurrence risk are typically around 1%. Genetic counselling with a clinical genetics service is recommended for families to discuss these issues and reproductive options. Prenatal genetic testing (chorionic villus sampling or amniocentesis) can detect known familial MECP2 mutations when desired.
In very rare cases a mother may be an asymptomatic or mildly affected carrier because of favourable X‑inactivation; such carriers have a 50% chance of passing the mutation to each child.
Around 5–10% of individuals with Rett‑like clinical features do not have MECP2 mutations. Other genes are implicated in variant forms that were previously classified within the Rett spectrum but are now recognised as distinct conditions:
Genetic counselling: Referral to clinical genetics is advised following diagnosis or strong clinical suspicion. Genetic counselling explains inheritance patterns, recurrence risks, testing options and reproductive choices and helps families understand implications for other relatives.
There is no known method to prevent most cases of Rett Syndrome since the causative mutations typically occur spontaneously. When a familial mutation is identified, prenatal testing can detect the variant in a pregnancy; prospective parents should discuss options with genetic services.
Rett Syndrome affects approximately 1 in 10,000 female live births worldwide. Prevalence appears similar across ethnic groups. In the United Kingdom this equates to an estimated 50–60 new diagnoses per year. Because many male embryos with pathogenic MECP2 variants do not survive to term and surviving males may present atypically, accurate prevalence estimates for males are uncertain.

Diagnosis of Rett Syndrome relies on careful clinical assessment of the developmental history and observed features, together with targeted genetic testing. Early signs overlap with other neurodevelopmental conditions, so referral for specialist assessment (typically via the general practitioner to paediatric neurology or clinical genetics) is recommended when developmental regression or characteristic features are identified.
Rett Syndrome is primarily a clinical diagnosis; recognition of a typical developmental trajectory and specific signs guides initial identification. The revised international diagnostic criteria published in 2010 remain the standard for classifying classic and atypical presentations.
An atypical or variant diagnosis is considered when at least two of the four core criteria plus five of the supportive criteria are present, typically following a period of regression and subsequent stabilisation.
Genetic testing is used to confirm the clinical impression. Analysis of DNA from a blood sample or other tissue identifies pathogenic variants in the MECP2 gene in the majority of individuals meeting criteria for classic Rett Syndrome (reported detection rates are around 90–95%). Testing also informs prognosis, family counselling and eligibility for mutation‑specific research studies and clinical trials.
Testing for alternative genes, such as CDKL5 and FOXG1, is appropriate when the clinical picture suggests these variant forms or when MECP2 testing is negative despite a strong clinical suspicion.
Not all individuals with MECP2 variants have a clinical diagnosis of Rett Syndrome, and not all cases with Rett‑like features have identifiable MECP2 mutations; the diagnosis rests on integrating clinical and genetic information.
Early-stage Rett Syndrome may be confused with other conditions. Key differentials include:
Assessment by clinicians experienced in neurodevelopmental disorders and access to multidisciplinary evaluation improves diagnostic accuracy.
Following identification of its genetic basis, Rett Syndrome is no longer classified as a primary psychiatric disorder in DSM‑5 but is recognised as a genetic neurodevelopmental condition. In ICD‑10 the condition appears under code F84.2 (“Rett’s syndrome”); ICD‑11 provides updated clarification of neurodevelopmental disorders with genetic origins.
Early and accurate diagnosis enables timely access to specialist interventions, coordinated multidisciplinary care and genetic counselling. Families benefit from prompt connection with support services, tailored therapies and information about clinical trials and research. Referral to paediatric neurology and clinical genetics should be considered when regression or core diagnostic features are suspected.
Connect with UK‑based organisations specialising in Rett Syndrome for guidance on diagnostic pathways and specialist clinics.
 No cure currently exists for Rett Syndrome. A coordinated, multidisciplinary treatment approach nevertheless meaningfully improves quality of life, maximises function and reduces secondary complications. Management emphasises symptomatic care, rehabilitation, prevention of complications and access to appropriate clinical trials or investigational therapies where available.
No cure currently exists for Rett Syndrome. A coordinated, multidisciplinary treatment approach nevertheless meaningfully improves quality of life, maximises function and reduces secondary complications. Management emphasises symptomatic care, rehabilitation, prevention of complications and access to appropriate clinical trials or investigational therapies where available.
At present, Rett Syndrome is not curable. Nevertheless, advances in understanding the underlying biology, notably the role of MECP2, have identified clear therapeutic targets. Preclinical studies in animal models have demonstrated that restoring functional MECP2 expression can reverse some features, supporting the rationale for future disease‑modifying approaches such as gene therapy and molecular treatments. Ongoing research aims to translate these findings safely into human trials.
Trofinetide (brand name Daybue) received US Food and Drug Administration approval in March 2023 as the first medication specifically indicated for Rett Syndrome in people aged two years and older. Trofinetide is an oral synthetic analogue of a naturally occurring neuropeptide and has shown modest improvements in clinical trial measures of core symptoms, including communication and hand function. Common adverse effects reported include diarrhoea and vomiting.
As of 2024 trofinetide is licensed in the United States and Canada; UK availability depends on regulatory review by the MHRA and subsequent appraisal by NICE. Families in the UK should discuss access pathways, compassionate use programmes and trial opportunities with their specialist team.
Several investigational therapies are under evaluation in clinical and preclinical studies:
Participation in clinical trials provides access to novel treatments and contributes to scientific progress. Families seeking information about current UK and international trials should consult specialist centres and reputable registries such as ClinicalTrials.gov or contact research charities like Reverse Rett.
Symptomatic care is individualised and delivered by a multidisciplinary team tailored to the person’s needs. Typical team members include:
Manages neurological issues, prescribes anticonvulsants and coordinates referrals to other specialists.
Provides programmes to maintain joint range, strengthen muscles, support mobility and reduce contractures.
Advises on adaptive equipment, hand function and activities of daily living.
Assesses swallowing safety and implements augmentative and alternative communication (AAC) strategies.
Manages nutritional needs, growth monitoring and feeding strategies, including gastrostomy where indicated.
Reviews and treats scoliosis and skeletal complications that may require surgical intervention.
Addresses reflux, constipation and motility problems.
Monitors cardiac rhythm and manages arrhythmic risks such as prolonged QT interval.
Supports behavioural management, mental health and family wellbeing.
Medication is used to treat specific symptoms and complications. Choice is individualised according to clinical presentation and tolerability:
Physiotherapy is central to management. Regular, goal‑directed therapy helps preserve mobility, maintain joint range, support posture and reduce the risk of contractures and scoliosis. Programmes commonly include standing practice, gait training, strength and balance work, hydrotherapy and respiratory physiotherapy.

Many individuals with Rett Syndrome struggle to maintain correct foot positioning during therapeutic exercises, particularly during sit-to-stand transitions and standing balance activities. Poor foot placement can compromise treatment effectiveness and require additional physical assistance from therapists or caregivers.
Clinical teams have observed positive outcomes with aids such as StandSure Therapy Aid, a rehabilitation device designed specifically to address these challenges. StandSure provides secure foot positioning through adjustable ankle retainers and toe straps, effectively anchoring feet flat on the footplate during weight-bearing exercises.
The device supports postural alignment during sit-to-stand transitions, enabling individuals to practice functional movements with proper foot placement. This allows therapists and families to focus on upper body support and trunk control without simultaneously managing foot positioning, which can be particularly beneficial when working with a single carer rather than requiring two-handed assistance.
StandSure is portable and available in multiple sizes to accommodate children and adults. Its ergonomic design fits between chair legs and wheelchairs, making it suitable for use in clinic, school, and home settings. For detailed information on clinical applications and sizing guidance, families and therapists can visit the How It’s Used page, which demonstrates various therapeutic exercises and positioning strategies.
As with any therapeutic equipment, StandSure should be used as part of a structured, supervised rehabilitation programme developed by qualified physiotherapists familiar with the individual’s specific needs and capabilities.
Feeding and swallowing difficulties are common and require individualised nutritional plans. Strategies include energy‑dense diets, texture modification, enteral feeding via gastrostomy when oral intake is insufficient, and regular monitoring of growth and nutritional biomarkers. Speech and language therapy assessment of swallowing safety is essential to reduce aspiration risk.
Scoliosis is frequent and may progress rapidly during growth. Management depends on curve severity and the individual’s overall health:
Early detection through routine spinal assessment and multidisciplinary planning is recommended.
Individuals with Rett Syndrome have an increased risk of cardiac arrhythmias, including prolonged QT interval. Recommended surveillance commonly includes baseline ECG at diagnosis, periodic ECG monitoring (often annually), Holter monitoring if symptoms or arrhythmias are suspected, and echocardiography where structural concerns exist. Avoidance of QT‑prolonging medications and cardiology review for documented abnormalities are important safety measures.
Educational programmes should be individually designed to support learning and communication. Augmentative and alternative communication (AAC), including eye‑gaze systems, can enable meaningful interaction. Special educational needs provision, sensory therapies, music therapy and behavioural strategies support participation and emotional wellbeing.
Sensory resources and adapted activities provide stimulation, enjoyment and therapeutic benefit. Occupational therapists can advise on appropriate toys and equipment, such as tactile items, musical aids, weighted products and visual sensory equipment.
Rett Syndrome places considerable demands on families and carers. Access to psychosocial support is essential and may include family therapy, counselling, respite care, peer support groups, assistance with care coordination, benefits advice and palliative care where appropriate. Clinicians should signpost families to national and local support organisations for practical help and community connection.

Living with Rett Syndrome presents substantial challenges, but with coordinated care many individuals achieve a meaningful quality of life. Families frequently report that their relatives remain emotionally present, affectionate and capable of expressing preferences despite severe physical limitations.
Many people with Rett Syndrome survive into adulthood. Life expectancy varies widely and depends on symptom severity, complication burden and access to comprehensive multidisciplinary care. Some individuals live into their 40s, 50s or beyond, while others experience life‑limiting complications earlier in life.
Factors that influence prognosis include:
Sudden unexpected death has been reported in this population and may relate to cardiac arrhythmias, brainstem dysfunction or respiratory events. Regular cardiac and respiratory surveillance, together with proactive management of seizures and nutritional status, are important measures to reduce risk.
Accurate assessment of intellectual capacity is difficult because severe motor and communication impairments limit standard testing methods. Conventional cognitive tests are often invalid for people who cannot speak or use their hands purposefully.
Clinical observations and family reports indicate that cognitive awareness and emotional responsiveness are frequently greater than physical abilities suggest. Many individuals demonstrate:
Modern eye‑gaze tracking and adapted communication technologies have revealed abilities previously underestimated. Clinical teams should presuppose competence and prioritise access to communication assessment and AAC interventions early after diagnosis.
Loss of spoken language does not preclude meaningful communication. Augmentative and alternative communication (AAC) approaches commonly used include:
Speech and language therapists with expertise in AAC should assess individual capability and recommend a personalised communication plan to maximise social participation and choice.
The butterfly has become an internationally recognised emblem of the Rett community. It symbolises transformation, resilience and silent communication—qualities that many families and advocacy groups associate with the condition. The symbol appears widely in awareness campaigns and fundraising activity led by organisations such as the International Rett Syndrome Foundation and Reverse Rett. Purple is commonly used for Rett awareness events, including annual Purple Day activities.
Austrian paediatrician Andreas Rett first described the syndrome in 1966. Subsequent publications, notably by Bengt Hagberg in the early 1980s, brought the condition to wider attention and the eponym “Rett syndrome” entered international use. The genetic basis was identified in 1999 when Huda Zoghbi and colleagues linked most cases to mutations in the MECP2 gene, a discovery that transformed research and clinical practice.
Families encounter distinct challenges at each life stage and benefit from proactive planning.
Focus is on early intervention, establishing diagnosis, initiating therapies and connecting with specialist services and peer support while adjusting to changed developmental expectations.
Attention turns to educational placement, development of communication systems, management of seizures and scoliosis, and promoting inclusion and social opportunities.
Transition planning includes adult health services, residential and social care options, continued therapy provision and legal matters such as lasting power of attorney and advance care planning.
As people with Rett Syndrome age, musculoskeletal complications, mobility decline and age‑related health conditions may emerge. Care plans should adapt to changing needs, integrating geriatric principles with lifelong disability care.
Families benefit from peer support, shared practical advice and local resources. Joining UK support groups can provide connection, information on services and emotional support.
 Families affected by Rett Syndrome in the United Kingdom can access dedicated charities and support networks that provide specialist information, practical assistance and links to research. These organisations offer helplines, regional groups, therapy advice and guidance on clinical trials, helping families navigate healthcare, education and social‑care systems.
Families affected by Rett Syndrome in the United Kingdom can access dedicated charities and support networks that provide specialist information, practical assistance and links to research. These organisations offer helplines, regional groups, therapy advice and guidance on clinical trials, helping families navigate healthcare, education and social‑care systems.
Website: www.rettuk.org
Telephone: 01582 798911
Email: [email protected]
Rett UK is the national charity offering information, advocacy and a helpline for families across the UK. Services include practical guides, regional support groups, family events and professional awareness work to improve local services and access to therapies.
Website: www.reverserett.org.uk
Telephone: 0161 5525001
Email: [email protected]
Reverse Rett focuses on research and drug development, funding studies into gene therapy, novel pharmaceuticals and clinical trials. The charity provides updates on ongoing trials and supports families seeking research participation and information on emerging treatments.
Website: www.brainwave.org.uk
Telephone: 01278 429089
Email: [email protected]
Brainwave provides specialist therapy programmes for children and young people with disabilities, including intensive assessments, personalised therapy plans and provision of adaptive equipment to support physical, communication and sensory needs.
Peer support offers practical advice and emotional connection. UK families often find timely, lived‑experience information through active online communities:
Website: www.facebook.com/rettsyndromesupport
This international group brings together families and carers to share practical tips, experiences of services and support around the world.
Website: www.facebook.com/rettsyndromesupportuk
This UK‑focused group concentrates on navigating the NHS, local services, benefits and country‑specific resources, providing a useful source of practical, locally relevant information.
There is no established method to prevent most cases of Rett Syndrome because the condition usually results from spontaneous genetic mutations. These de novo mutations occur randomly during formation of parental germ cells or early embryonic development and are not caused by parental actions or environmental exposures. When a familial pathogenic variant is known, prenatal testing (chorionic villus sampling or amniocentesis) can detect the variant in a pregnancy; families with an affected child should be offered genetic counselling to discuss recurrence risk and reproductive options.
Rett Syndrome in males is uncommon but can occur under particular circumstances. Survival with Rett‑like features is possible in males who have Klinefelter syndrome (47,XXY), in males with somatic mosaicism for an MECP2 mutation, or when the mutation is particularly mild. Males may also present with other MECP2‑related disorders such as severe neonatal encephalopathy; without protective factors many male embryos with pathogenic MECP2 variants do not survive to term.
Rett Syndrome is caused by pathogenic variants in genes such as MECP2 (most commonly) that are located on the X chromosome. In the great majority of cases the mutation arises de novo in the parental germline rather than being inherited from a clinically affected parent. A small proportion of cases result from an unaffected mother who is a carrier due to favourable X‑inactivation. The condition follows an X‑linked dominant pattern, which explains its predominance in females.
There is currently no cure for Rett Syndrome. Research has, however, identified promising therapeutic targets. Preclinical studies show that restoring functional MECP2 can reverse some changes in animal models, and several investigational approaches — including gene therapy, read‑through compounds and small molecules — are under study. Trofinetide became the first drug specifically approved for Rett Syndrome by the US FDA in 2023; it offers modest symptomatic benefit but is not curative. Families interested in experimental treatments should discuss clinical‑trial options with their specialist centre.
Life expectancy in Rett Syndrome varies considerably. Many individuals live well into adulthood, with some reaching their 40s, 50s or beyond. Prognosis depends on multiple factors including the specific MECP2 mutation, seizure control, respiratory and cardiac health, nutritional status and access to multidisciplinary care. Reports of sudden unexpected death exist; clinicians therefore recommend regular cardiac and respiratory surveillance and proactive management of complications to optimise longevity and quality of life.
Rett Syndrome is distinct from autism spectrum disorder, though there is clinical overlap. Historically it was classified with pervasive developmental disorders, but following discovery of its genetic basis; it is recognised as a genetic neurodevelopmental condition. Key distinguishing features include a period of apparently normal early development followed by regression, characteristic hand stereotypies and breathing irregularities. Some autistic‑like behaviours may appear transiently during regression.
Trofinetide (Daybue) is the first medication specifically licensed for Rett Syndrome by the US FDA in March 2023 for use in people aged two years and older. It is an oral analogue of a neuropeptide and has demonstrated modest improvements in clinical trials. As of 2024, trofinetide is available in the United States and Canada; UK availability depends on MHRA licensing and NICE appraisal. Families should consult their specialist care teams about local access, compassionate use schemes and clinical‑trial options.
Rett Syndrome is considered a neurodevelopmental rather than a primarily degenerative disorder. Neurones are present but show atypical maturation and connectivity rather than progressive cell loss. While motor function may decline in later years due to musculoskeletal complications, the underlying pathology differs from classic neurodegenerative diseases.
Multiple avenues are under investigation, including gene therapy to restore MECP2 function, read‑through compounds for nonsense mutations, small molecules such as blarcamesine (Anavex 2‑73), protein‑replacement strategies and drugs that modulate downstream pathways. ClinicalTrials.gov and charities such as Reverse Rett and Rett UK provide current trial listings and information; discuss eligibility and risks with a specialist centre before considering participation.
The butterfly has become a widely recognised symbol for the Rett community, representing transformation, resilience and non‑verbal communication. It features in awareness campaigns and fundraising activities, often accompanied by the colour purple on events such as Purple Day for Rett.
 Rett Syndrome is a complex neurodevelopmental condition that substantially affects communication, motor function and daily living. Despite these challenges, many individuals demonstrate emotional presence and capacity for meaningful interaction, and families frequently describe rich, affectionate relationships with their relatives.
Rett Syndrome is a complex neurodevelopmental condition that substantially affects communication, motor function and daily living. Despite these challenges, many individuals demonstrate emotional presence and capacity for meaningful interaction, and families frequently describe rich, affectionate relationships with their relatives.
Knowledge of the genetic basis, clinical progression and evidence‑based management enables families and clinicians to plan care that optimises quality of life. A multidisciplinary approach — incorporating neurology, physiotherapy, nutrition, orthopaedics and psychological support — addresses the varied needs of people with Rett Syndrome throughout life.
Research continues to progress. The identification of MECP2 in 1999 provided a clear target for experimental therapies, and the approval of trofinetide in 2023 represented a milestone as the first disease‑specific medication. Ongoing studies into gene therapy, small molecules and other molecular approaches offer potential for future disease‑modifying treatments, but expectations should remain measured until safety and efficacy are established in clinical trials.
Access to support services is essential. UK organisations such as Rett UK, Reverse Rett and Brainwave provide practical information, peer connection and pathways to research involvement. Clinicians should signpost families to reputable charities and trial registries and discuss trial eligibility via specialist centres.
In clinical practice, early recognition, timely referral and coordinated, compassionate care improve outcomes and family experience. Physiotherapy and assistive aids help maintain function; regular cardiac and respiratory surveillance, nutritional management and seizure control reduce complication risks. Above all, care should preserve dignity, promote participation and support family wellbeing.
Collectively, through continued research, improved clinical care and strong community support, the outlook for individuals with Rett Syndrome continues to improve. Families and clinicians alike benefit from accurate information, practical resources and the shared hope that future advances will further enhance lives.
Book a personalised demonstration of the StandSure™ today by filling out our request form. Have any questions? Call us now, and our team will be happy to assist!



