No products in the cart.

Medical Disclaimer: This article details educational information about muscular dystrophy. Any individuals with muscle weakness should see their GP or consult NHS 111 to have their condition professionally assessed. The sooner a diagnosis is made the sooner care can be appropriately planned.
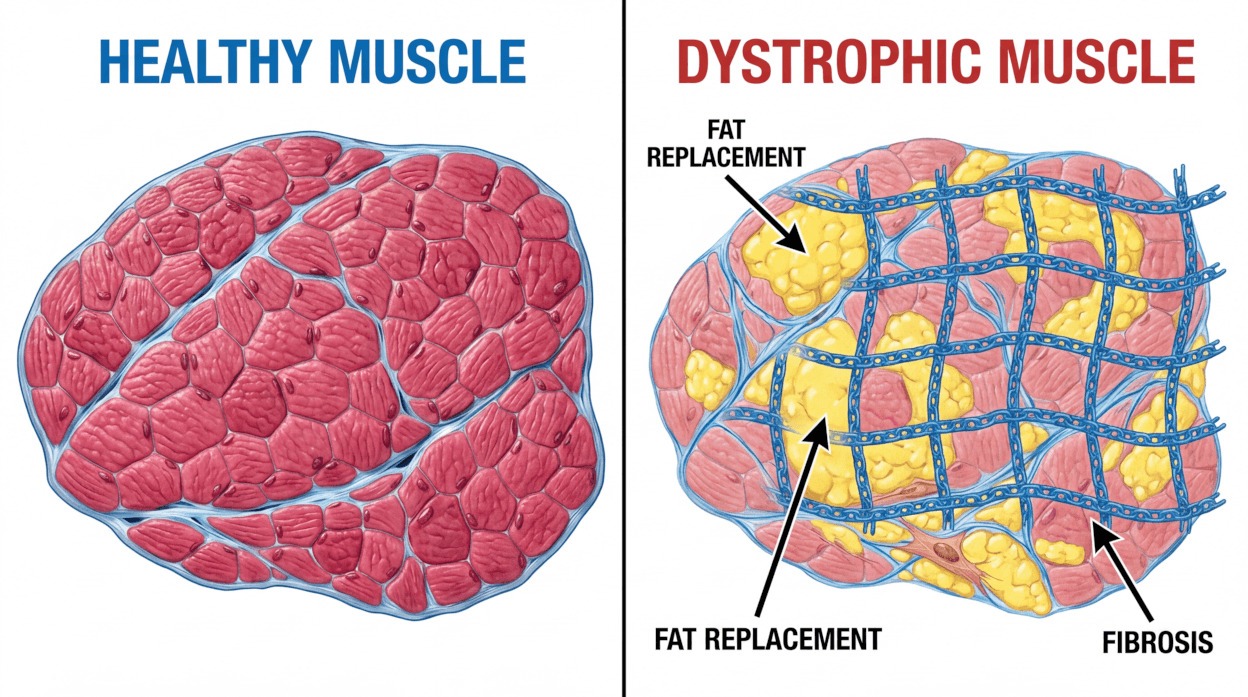
Muscular dystrophy refers to more than 60 genetically distinct conditions that impair muscle function. The term ‘dystrophy’ denotes abnormal development or degeneration of tissue; in this context, muscle fibres progressively weaken and break down, causing increasing disability.
Each type results from specific gene mutations that reduce or alter production of proteins essential for muscle structure and function. Without these proteins, muscles lose strength and structural integrity over time.
 The clinical course varies by type. Some people experience rapid progression with early loss of mobility, while others maintain good function into adulthood with appropriate management. Weakness commonly begins in particular muscle groups and then spreads; recognising the pattern (for example, proximal versus distal weakness) assists clinicians in identifying the likely type.
The clinical course varies by type. Some people experience rapid progression with early loss of mobility, while others maintain good function into adulthood with appropriate management. Weakness commonly begins in particular muscle groups and then spreads; recognising the pattern (for example, proximal versus distal weakness) assists clinicians in identifying the likely type.
Patterns of weakness and system involvement help guide diagnosis, monitoring and treatment planning. Cardiac and respiratory muscles are affected in several types, so multidisciplinary assessment is essential.
Early recognition of muscular dystrophy symptoms enables prompt assessment and onwards management. Presentation varies by type and age of onset; symptoms most often develop gradually, although some forms can present more suddenly.
Muscle weakness is the cardinal feature across the spectrum of dystrophies. The distribution of weakness differs by type and helps guide diagnosis. Developmental delays in young children commonly prompt assessment, while adults may notice progressive loss of strength during routine activities.

Parents and carers may notice several early indicators of muscular dystrophy. Delayed motor milestones — for example, later walking — and an unusual gait pattern often provide the first clue.
Frequent falls and difficulty rising from sitting or lying positions are common as proximal hip and thigh muscles weaken. Some children use their hands to “walk up” their legs when standing; this is known as Gower’s manoeuvre and is characteristic of certain types (most notably Duchenne).
Adult-onset muscular dystrophy often progresses more slowly, permitting gradual adaptation. Weakness may begin in the face, shoulders or hips and spread over time.
Common adult symptoms include reduced endurance for sustained tasks, difficulty lifting the arms, and diminished grip strength. Involvement of bulbar or respiratory muscles can cause swallowing or breathing symptoms.
Certain developments require immediate medical attention. New or worsening breathing difficulties may indicate respiratory muscle involvement and need urgent assessment. Severe chest pain, fainting, sudden loss of mobility or difficulty swallowing also warrant prompt evaluation to prevent complications.
Seek Immediate Medical Help If: Sudden breathing difficulties, chest pain, severe muscle pain, loss of mobility, or difficulty swallowing occurs. These symptoms may indicate disease progression requiring urgent medical management.
Progression rates differ between types. Regular clinical review allows monitoring of muscle weakness and early detection of cardiac or respiratory complications so that treatment can be adjusted promptly and quality of life maintained.
Muscular Dystrophy UK provides expert information and emotional support for families navigating diagnosis and symptoms. Their helpline connects you with specialists who understand your concerns.
Muscular dystrophy results from gene mutations that impair production of proteins essential for normal muscle structure and function. These genetic changes determine the specific type of dystrophy, explain patterns of inheritance within families, and inform risk assessment and reproductive counselling.
Many muscular dystrophies arise when a vital muscle protein is missing or dysfunctional. Dystrophin, for example, stabilises muscle cells during contraction and relaxation; when dystrophin is absent or markedly reduced, muscle fibres become fragile and progressively deteriorate.
Different types follow distinct inheritance patterns, which influence who is at risk and what testing is appropriate.
X‑linked recessive inheritance is responsible for the most common childhood forms, notably Duchenne and Becker muscular dystrophy. In X‑linked conditions, carrier mothers may be asymptomatic, while affected boys typically develop the disease.
Autosomal dominant inheritance requires a single mutated copy of a gene from either parent to cause disease. Each child of an affected parent has around a 50% chance of inheriting the mutation. Facioscapulohumeral muscular dystrophy commonly follows this pattern.
Autosomal recessive inheritance requires two mutated copies, one from each parent. Carriers with a single mutated copy are usually asymptomatic. Many limb‑girdle muscular dystrophy subtypes follow autosomal recessive transmission.
Mutations in the DMD gene cause Duchenne muscular dystrophy, the most severe childhood form. The DMD gene normally produces dystrophin; absence of functional dystrophin leads to rapid muscle decline in affected boys.
Becker muscular dystrophy is caused by different mutations in the same DMD gene that permit some dystrophin production, resulting in a milder, later‑onset phenotype than Duchenne.
Myotonic dystrophy arises from a trinucleotide repeat expansion, an unusual genetic mechanism in which sections of DNA are abnormally repeated. Longer repeat lengths generally associate with earlier onset and more severe features.
Genetic counselling supports families by explaining inheritance patterns, outlining testing options and discussing reproductive choices. Counsellors advise on carrier testing, prenatal diagnosis and preimplantation genetic diagnosis where appropriate.
Genetic testing identifies the precise mutation, confirms the diagnosis and informs prognosis and eligibility for mutation‑specific treatments or trials.
Not all cases result from inherited changes. De novo (spontaneous) mutations can arise in the egg, sperm or early embryo, leading to disease in a child with no prior family history. Such mutations may subsequently be transmitted to later generations.
Estimates indicate a substantial proportion of Duchenne cases arise from de novo mutations; precise figures vary by study and should be verified with current genetic data during counselling.
Medical science recognises more than 30 distinct types of muscular dystrophy. Each type targets particular muscle groups, progresses at a characteristic rate and presents at a typical age. Understanding these differences helps clinicians tailor monitoring and treatment to individual needs.
Classification considers genetic cause, inheritance pattern, age of onset and the primary muscles affected. Some types appear in childhood, while others present in adolescence or adulthood. Severity ranges from mild weakness to profound impairment.
Duchenne muscular dystrophy is the most common and severe childhood form. It affects mainly boys and typically presents between ages two and five. Muscle weakness usually begins in the hips and thighs, then spreads to the shoulders and upper arms.
The condition progresses relatively rapidly; many affected boys require a wheelchair in adolescence without treatment. Cardiac and respiratory muscles are commonly involved as the disease advances. Duchenne results from mutations in the DMD gene that prevent production of functional dystrophin, a protein essential for muscle cell stability.
Becker muscular dystrophy shares the same gene as Duchenne but follows a milder course. Symptoms generally begin later, often in adolescence or early adulthood, and progression is slower. Mutations permit some dystrophin production, which partially preserves muscle function.
Cardiac involvement can occur and requires routine assessment. Many people with Becker maintain ambulation into middle adulthood.
Myotonic dystrophy is the commonest adult‑onset muscular dystrophy, though congenital forms exist. It affects approximately one in 8,000 people and comprises two main forms, DM1 and DM2, caused by different genetic mechanisms.
Myotonia (delayed muscle relaxation) characterises the condition. Weakness often affects the face, neck and distal limbs first. Multisystem features include cardiac conduction abnormalities, cataracts and endocrine or cognitive problems, requiring coordinated care across specialities.
Facioscapulohumeral muscular dystrophy (FSHD) commonly begins in the teenage years or early adulthood. Weakness typically affects the face, shoulder blades and upper arms and is often asymmetric. Progression is usually slow and many people retain mobility.
Limb‑girdle muscular dystrophy (LGMD) describes a group of over 30 subtypes caused by a range of genetic mutations. These predominantly affect the muscles around the hips and shoulders. Age of onset and severity vary by subtype, and both autosomal dominant and recessive inheritance patterns occur.
Certain LGMD subtypes involve cardiac or respiratory muscles and therefore require specialist surveillance and management.
Congenital muscular dystrophy presents at birth or in early infancy. Babies may show hypotonia, delayed motor development or joint contractures. Some congenital types are associated with brain abnormalities affecting development; others predominantly involve muscle. Severe cases may need early respiratory support and intensive physiotherapy to optimise outcomes.
A rarer form notable for early joint contractures (elbows and ankles) and slowly progressive weakness of upper arms and lower legs. Cardiac conduction disease is common and often necessitates specialist cardiology review; pacemaker insertion may be required in some individuals.
Typically presenting after age 40, this adult‑onset type causes drooping eyelids (ptosis) and swallowing difficulties. Weakness may extend to shoulder and hip muscles over time.
These rare variants predominantly affect muscles furthest from the trunk — hands, feet, lower arms and lower legs. Onset is usually in adulthood and progression is generally slow.
Accurate diagnosis requires a staged evaluation combining clinical assessment, laboratory tests and genetic analysis. The diagnostic pathway aims to identify the specific type of muscular dystrophy so that treatment, monitoring and genetic counselling can be tailored to the individual.
Initial assessment commonly begins when symptoms prompt medical review. Primary‑care clinicians examine muscle strength, reflexes and movement patterns and take a detailed family history to identify potential inherited patterns. Blood tests, particularly creatine kinase (CK), often provide the first biochemical indication of muscle damage.
Raised CK levels suggest muscle injury but do not identify the cause. Electromyography (EMG) assesses muscle electrical activity and helps distinguish myopathic from neurogenic processes. Muscle biopsy, when performed, provides histological and immunohistochemical information.
Genetic testing is definitive for most types. DNA analysis from a blood sample detects characteristic gene changes, confirming the diagnosis and specifying the mutated gene or repeat expansion involved. Identification of the causative gene informs prognosis and eligibility for mutation‑specific treatments or trials.
Baseline cardiac assessment with electrocardiography (ECG) and echocardiography, plus pulmonary function tests, are important parts of initial evaluation. These establish a monitoring schedule to detect early cardiac or respiratory involvement.
Age at diagnosis varies by type. Duchenne muscular dystrophy is commonly diagnosed between three and five years, often after delayed motor milestones are noted. Some regions offer newborn screening pilots, which can detect certain disorders earlier where available.
Becker muscular dystrophy frequently presents in adolescence or early adulthood, while adult‑onset types such as myotonic dystrophy or facioscapulohumeral muscular dystrophy may not be identified until middle age. Congenital types are recognised at birth or in early infancy.
Early and accurate diagnosis enables timely interventions that may slow progression and reduce complications. Referral to a regional neuromuscular centre is recommended for diagnostic confirmation, multidisciplinary management and discussion of genetic testing and family implications—particularly if there is rapid progression or cardiac/respiratory symptoms.
Specialist Referral: Diagnosis and ongoing management typically involve neuromuscular specialists at regional centres. These multidisciplinary teams include neurologists, geneticists, physiotherapists, and other specialists working collaboratively to optimise care.
Although no cure exists at present, comprehensive treatment substantially improves quality of life and life expectancy for many people with muscular dystrophy. Management aims to slow progression, preserve function, prevent complications and support independence. Care is delivered by multidisciplinary teams coordinating across relevant specialities.
Treatment is individualised according to the specific type of dystrophy, current symptoms and rate of progression. Regular review allows timely adjustment of therapies. Typical approaches combine medication, physiotherapy, assistive devices and, when indicated, surgical intervention.

Corticosteroids are the established medical therapy for Duchenne muscular dystrophy. Prednisolone and deflazacort are commonly used in the UK and have been shown to slow muscle deterioration and preserve strength for several years.
Therapy often commences before significant loss of mobility to maximise benefit. Corticosteroids also reduce the risk of progressive scoliosis and delay respiratory complications. Side effects can include weight gain, mood changes, reduced bone density and growth suppression; these require active monitoring and management by the clinical team.
Advances in genetic treatments provide mutation‑specific options for selected patients. Exon‑skipping therapies enable cells to produce shortened but partially functional dystrophin in people with particular DMD gene mutations. A minority of Duchenne patients are eligible for current exon‑skipping approaches; eligibility depends on the precise genetic change.
Gene therapy trials aim to deliver functional gene copies to muscle tissue. Early results are encouraging, but long‑term safety and effectiveness require further study. Regulatory approvals are expanding gradually for specific patient groups.
Cardiac muscle involvement occurs in several dystrophy types and requires proactive management. Angiotensin‑converting enzyme (ACE) inhibitors and beta‑blockers help preserve cardiac function and delay cardiomyopathy. Regular echocardiography and ECG monitoring detect developing problems early.
Some individuals require devices such as pacemakers or implantable cardioverter‑defibrillators (ICDs) to manage rhythm disturbances. Cardiologists and neuromuscular specialists coordinate ongoing cardiac care.
Respiratory muscle weakness necessitates timely respiratory intervention in progressive forms. Non‑invasive ventilation (NIV) commonly provides nocturnal support and may progress to daytime use as needs evolve. Regular pulmonary function testing informs timing of interventions.
Cough assist devices and respiratory physiotherapy techniques improve airway clearance and reduce infection risk. Early respiratory support has a significant favourable effect on life expectancy and quality of life.
Orthopaedic procedures address complications such as scoliosis and joint contractures. Spinal fusion can stabilise severe curvature, improving sitting balance and potentially supporting respiratory function. Tendon releases may increase range of motion when contractures limit activity.
Timing of surgery is individual and requires careful consideration of benefits and risks, including peri‑operative cardiac and respiratory assessment.
Treatment Advances: Ongoing research investigates gene editing, stem cell approaches and muscle‑regenerating compounds. Clinical trials provide access to experimental therapies for eligible participants. Further information about current studies and trial eligibility is available from specialist neuromuscular centres and Muscular Dystrophy UK.
Medication supports both disease modification and symptom control in muscular dystrophy. Choice of drug therapy depends on the confirmed genetic diagnosis, current clinical features and individual tolerance. Close specialist supervision is essential for any disease‑modifying treatment.
Beyond therapies aimed at the underlying genetic defect, medications treat secondary problems that affect quality of life. Common targets include pain, muscle cramps, sleep disturbance and mood changes. A balanced regimen seeks to control symptoms while minimising adverse effects.
Corticosteroids remain the principal disease‑modifying medication for Duchenne muscular dystrophy; prednisolone and deflazacort are commonly prescribed in the UK. These agents slow muscle deterioration and prolong functional ability, but require regular monitoring for side effects.
Mutation‑specific therapies are available for selected patients. Ataluren is an oral treatment for certain nonsense mutations in the DMD gene. Other antisense oligonucleotide or exon‑skipping agents (for example eteplirsen, golodirsen, casimersen and viltolarsen) target specific exons of the DMD gene to enable production of partially functional dystrophin. Eligibility depends on precise genetic testing and specialist assessment.
When the heart is affected, angiotensin‑converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) are used to reduce cardiac workload and delay cardiomyopathy. Beta‑blockers may be added to manage heart rate and rhythm. Diuretics and specific antiarrhythmic drugs are used as clinically indicated.
Muscle cramps and neuropathic pain respond to a range of options. Magnesium supplements can reduce cramp frequency for some people; gabapentin or carbamazepine may help neuropathic pain. Non‑steroidal anti‑inflammatory drugs or other analgesics are used for musculoskeletal discomfort related to altered biomechanics.
Sleep problems are common; melatonin or other sleep‑regulating approaches may be beneficial. Treating pain, breathing difficulties or restless legs often improves sleep quality. Constipation is frequently encountered and is managed with dietary measures, stool softeners, laxatives or motility agents as required.
Gene therapy products and single‑administration vector‑based treatments are under regulatory review and in clinical trials; these aim to provide lasting benefit by delivering functional gene copies to muscle tissue. Anti‑myostatin agents, which target pathways that limit muscle growth, are also under investigation as adjunctive therapies.
All novel pharmaceutical approaches require specialist assessment, enrolment in appropriate clinical trials where available and careful long‑term follow‑up. Medication management should be coordinated between neuromuscular specialists, the GP and the pharmacist, and families should keep an up‑to‑date medication list and report new symptoms promptly.
Physiotherapy is a central component of care for people with muscular dystrophy across the disease course. Specialist interventions aim to maintain mobility, prevent complications and optimise functional ability. Physiotherapists collaborate with families to develop personalised, adaptable exercise and posture programmes.
Programmes balance activity with appropriate rest to avoid overexertion. Gentle stretching, targeted strengthening and mobility training help preserve muscle function while reducing the risk of contractures. Regular review ensures exercises remain suitable as muscle strength and movement change.

Regular physiotherapy can slow functional decline in muscular dystrophy. Stretching maintains joint range of motion and reduces the development of contractures that limit movement. Carefully graded strengthening preserves existing muscle function without causing damage from excessive strain.
Respiratory physiotherapy supports breathing by strengthening respiratory muscles and improving secretion clearance. Postural management reduces chest wall deformity that can compromise lung capacity, and breathing exercises form an integral part of long‑term care.
Standing programmes offer important musculoskeletal benefits for children as they lose independent mobility. Weight‑bearing activity supports bone density, helps maintain hip joint alignment and supports circulation and digestive function. These interventions remain valuable even when walking becomes limited.
Standing aids provide a practical means of maintaining safe, supported weight‑bearing. The StandSure Therapy Aid is one such device recommended by physiotherapists as part of a comprehensive management plan. It supports hands‑free standing and helps children participate in activities at standing height.
Standing sessions should be individualised and adjusted to tolerance; many programmes incorporate regular sessions integrated into daily routines at home or school settings to support bone health and joint alignment.

The StandSure device enables safe, supported standing for children with reduced mobility. Physiotherapists may incorporate this aid to help maintain lower limb alignment and bone health while children engage in standing‑level activities.
The equipment can be used at home or school. Session length and frequency should be tailored to the child’s tolerance and therapy goals.
Physiotherapists design individualised exercise plans that reflect current ability and long‑term goals. Low‑impact activities such as swimming provide cardiovascular conditioning with minimal muscle strain. Hydrotherapy combines exercise and relaxation and often benefits both physical and emotional wellbeing.
Home exercise programmes promote daily practice between supervised sessions. Simple stretches, gentle strengthening and functional activities maintain gains achieved in therapy. Family involvement supports adherence and enables integration into everyday life.
As the condition progresses, exercises are adapted to focus on maintaining function, preventing secondary complications and supporting comfort and participation rather than increasing strength.
Physiotherapists assess and recommend mobility aids and adaptive equipment to match current and anticipated needs. Manual wheelchairs may preserve walking for short distances, while powered wheelchairs provide independence when manual propulsion is no longer feasible.
Ankle‑foot orthoses (AFOs) support foot and ankle position to prolong walking ability. Spinal orthoses may help manage scoliosis progression in some individuals. Standing frames, including the StandSure device, continue to provide weight‑bearing benefits as mobility declines.
Equipment prescription considers present function and likely future needs. Regular reassessment, correct fitting and user training are essential to ensure safety and optimise benefit for movement, joints and daily activities.
Accessing Physiotherapy: NHS physiotherapy services provide ongoing support for muscular dystrophy management. Regional neuromuscular centres offer specialist physiotherapy with expertise in progressive conditions. Private physiotherapy options may complement NHS services where additional sessions are required.
Adapting to life with muscular dystrophy requires physical, emotional and practical adjustments. Well‑matched support systems help people and families maintain quality of life and continue participation in valued activities. Peer support and specialist information provide practical guidance and emotional reassurance.
Environmental modifications, assistive technology and care support maximise independence at home, school and work. Educational adjustments enable children to access learning; timely occupational and physiotherapy input helps optimise function and participation.

Muscular dystrophy affects many aspects of daily life. Mobility limitations can make self‑care, household tasks and recreational activities harder. Fatigue is common and often requires energy‑conservation strategies and careful planning of activities.
With anticipatory planning — accessible venues, suitable transport and appropriate equipment — social participation can be maintained. Regular review of needs and supports helps preserve independence as requirements change.
A progressive condition poses emotional challenges for individuals and families. Grief, anxiety and depression are not uncommon and benefit from timely psychological support. Specialist counselling or local mental‑health services can assist in developing coping strategies.
Children may experience difficulties with peer relationships and self‑image; integrated support that addresses both emotional development and physical needs promotes healthier adjustment. Family therapy or counselling may help manage changing roles and sibling relationships.
Schools can provide Education, Health and Care Plans (EHCPs) or equivalent personalised support to address mobility, fatigue and learning needs. Regular reviews ensure arrangements adapt to changing circumstances.
Further education and employment remain achievable with reasonable adjustments under the Equality Act 2010. Careers advice should consider physical capabilities alongside individual strengths and aspirations; workplace adaptations often enable continued employment.
Financial support in the UK includes benefits such as Personal Independence Payment (PIP) for adults and Disability Living Allowance (DLA) for children, where eligible. Carer’s Allowance may support unpaid family carers. Local authority social services assess care needs and can arrange direct payments or commissioned care.
NHS services and wheelchair or equipment services provide assessments and equipment to support daily living. Disabled Facilities Grants and other funding streams can assist with housing adaptations such as ramps, stairlifts or accessible bathroom facilities. Occupational therapists advise on environmental changes to improve safety and independence.
Specialist charities provide essential support for people and families affected by muscular dystrophy. These organisations offer reliable information, emotional support, practical assistance and fund research. Connecting with such services helps navigate practical challenges and builds supportive communities.
Support commonly includes telephone helplines, online forums, local groups and family events. Charities also fund research into improved treatments and advocate for services and policy that reflect the needs of the muscular dystrophy community.

The UK’s leading charity supporting more than 60 muscle‑wasting conditions. MDUK provides comprehensive information resources, funds research and offers practical support through helplines and local groups to benefit individuals at all stages.
Services: Information helpline, financial grants, support groups, research funding, advocacy
Address: 32 Ufford Street, London SE1 8QD
Monday to Friday, 10am–5pm
A charity focused on accelerating research and improving treatments for Duchenne muscular dystrophy. Duchenne UK funds studies and supports access to clinical trials for affected boys and men.
Focus: Research funding, clinical trial support, treatment access advocacy
Provides community support and information for families affected by Duchenne. The charity offers practical guidance and advocates for improvements in treatment and standards of care.
Services: Family support, information resources, community events, advocacy
Monday–Friday, 9am–5pm
Run by families for families, this group offers peer support and practical advice from experienced parents. Their helpline helps newly diagnosed families connect with those who understand the challenges.
Approach: Peer‑to‑peer support, practical guidance, emotional understanding
Provides condition‑specific support for people with myotonic dystrophy and their families. Services include specialist information, regional contacts and peer‑support networks.
Services: Helpline, regional groups, specialist information, annual conferences
Address: 19-21 Main Road, Gedling, Nottingham NG4 3HQ
An experiential charity focusing on children and young people; offers bespoke “Muscle Dreams” interventions designed to improve mental wellbeing, reduce isolation and build confidence.
Unique Offering: Personalised experiences, emotional wellbeing support, confidence building
Email: [email protected]
Offers free, ongoing specialist therapies for adults with neuromuscular conditions, including physiotherapy, osteopathy, exercise classes and hand therapy delivered by clinicians experienced in muscle‑wasting disorders.
Location: Unit 10, Westwood House, Westwood Way, Coventry CV4 8HS
Services: Physiotherapy, osteopathy, exercise programmes, hand therapy
Email: [email protected]
These charities offer complementary services alongside NHS care and often work collaboratively to meet different needs. Many families engage with multiple organisations to access the mix of information, emotional support and practical assistance most relevant to their circumstances.
Research continues to advance understanding of muscular dystrophy and to develop novel therapeutic approaches. Gene therapy, gene editing and strategies to regenerate or protect muscle tissue are active areas of investigation. Clinical trials test a range of interventions that may change outcomes for selected patient groups over time.
In recent years, progress has moved from laboratory discovery to treatments available for specific genetic subgroups, such as certain exon‑skipping therapies. Early gene therapy trials have reported encouraging safety and efficacy signals in carefully selected participants, but long‑term benefits and wider applicability require further study.
Gene editing technologies such as CRISPR offer the potential for precise correction of disease‑causing genes; at present, evidence derives primarily from preclinical and early‑phase human studies. Stem cell approaches aim to restore or replace damaged muscle tissue by delivering healthy precursor cells, while anti‑inflammatory strategies seek to limit secondary damage that contributes to progression.
Work also focuses on improving delivery methods, reducing immune responses to therapies and developing biomarkers to measure treatment effect. Progress is incremental and must be appraised in the context of robust clinical trial data and regulatory review.
Clinical trials and observational studies rely on volunteer participation to advance knowledge. Information about current studies is available via specialist neuromuscular centres, charity websites and recognised trial registries. Participation can provide access to experimental therapies but involves potential risks and burdens that should be discussed with the clinical team.
Observational studies that document the natural history of different types of muscular dystrophy are equally valuable; they inform trial design and outcome measures. Eligibility criteria vary by study and may specify age groups, genetic diagnosis or disease stage.
Hope Through Research: Progress in genetic and regenerative medicine offers cautious optimism. Whilst broadly applicable cures are not yet established, ongoing research and trial participation are essential steps towards more effective treatments for people living with muscular dystrophy.
Age at diagnosis depends on the specific type of muscular dystrophy. Duchenne muscular dystrophy is most often identified in early childhood, commonly between three and five years, when delayed motor milestones and muscle weakness become apparent. In some areas, newborn screening may detect particular disorders earlier where programmes exist.
Becker muscular dystrophy often presents later, during adolescence or early adulthood. Congenital types are recognised at birth or in early infancy because of reduced muscle tone or delayed development.
Adult‑onset types such as myotonic dystrophy or facioscapulohumeral muscular dystrophy may not be diagnosed until middle age or later. Gradual symptom onset or lack of family history can delay diagnosis in some people.
Muscular dystrophy affects many aspects of life beyond movement. Progressive muscle weakness reduces independence for self‑care tasks such as dressing and bathing and may eventually necessitate mobility aids or a wheelchair, depending on the type and progression.
Respiratory muscle weakness and cardiac involvement are serious complications in several types and require ongoing monitoring and treatment. Some forms affect swallowing, speech or vision, adding further care needs.
Social, educational and vocational participation may require adaptations. With appropriate medical management, assistive technology and support services, many people maintain fulfilling and productive lives despite physical limitations.
First signs vary by type and age. In childhood forms such as Duchenne, early indicators include delayed motor milestones (for example, late walking), difficulty running, frequent falls and problems climbing stairs. Difficulty rising from the floor is a common early sign.
Enlarged calf muscles (pseudohypertrophy) and use of the hands to “climb up” the legs when standing (Gower’s sign) are typical in Duchenne but not universal across all dystrophies.
In adult‑onset types, initial symptoms may be subtle weakness in specific muscle groups, facial weakness, difficulty lifting the arms or reduced grip strength. Some people notice muscle stiffness or cramping before clear weakness develops.
The progressive loss of muscle strength and the need for repeated adjustment to changing abilities are major challenges for individuals and families. Loss of independence and the emotional impact of progressive disability are central concerns.
Medically, respiratory complications are often the most serious issue in progressive forms, as weakened breathing muscles increase the risk of infections and can be life‑limiting without appropriate intervention. Cardiac disease is another key threat in types that affect the heart.
Practical barriers — such as inaccessible environments, transport difficulties and limited availability of specialised services — compound the burden. Comprehensive, multidisciplinary support helps address these medical and social challenges.
Pain varies between individuals and by type of dystrophy. The primary disease process does not typically cause severe pain for most people, but secondary problems commonly produce discomfort that requires management.
Muscle cramps, joint stiffness from contractures and musculoskeletal pain related to altered posture and movement patterns are frequent sources of symptoms. Scoliosis or prolonged sitting can cause back or pressure‑related pain.
Pain management strategies include appropriate medications, physiotherapy, positional adjustments and equipment to relieve pressure. A personalised approach from the multidisciplinary team usually controls most pain effectively.
Progression rates differ markedly between types. Duchenne muscular dystrophy generally progresses more rapidly; without corticosteroid treatment, many boys historically required a wheelchair by around ages 10–12, although treatments and supportive care have extended functional ability and life expectancy in recent years.
Becker muscular dystrophy typically advances more slowly, with many people remaining ambulant into their twenties or thirties. Adult‑onset types such as facioscapulohumeral muscular dystrophy often progress very gradually, and many individuals retain independence for years.
Myotonic dystrophy shows variable progression depending on the subtype. Individual factors — including the exact genetic mutation, treatment access, complication management and lifestyle — influence the rate of decline. Regular clinical monitoring allows timely adjustment of care as needs change.
Questions About Your Situation? Individual experiences with muscular dystrophy vary considerably. Specialist neuromuscular clinics provide personalised information addressing specific circumstances. Contact your healthcare team or charity helplines for guidance tailored to your needs.
Living with muscular dystrophy is challenging. Managing muscle dystrophy involves coordinated care, practical support, and emotional support. With the help of multidisciplinary teams, assistive technologies and community resources, flexible approaches help preserve the quality of life for many.
When early interventions are possible and detected earlier, the rate of progression and the number of potential complications are reduced. Cardiac, respiratory, orthopaedic and psychological issues should be managed proactively. Maintaining function and independence may be supported by physiotherapy, standing programmes or mobility aids such as the StandSure device.
Some of the most practical aid, reliable information and emotional support are provided by specialist charities. Their services are an important addition to clinical care as they provide peer support, advocacy, and many other things. Counselling and equipment guidance are also available.
Families affected by muscular dystrophy demonstrate resilience. As supportive networks, they offer guidance and encouragement to challenged families. Included healthcare professionals, charities and other families. Although challenges remain, there is growing optimism with the steady progress of research and care.
Collective efforts in research participation, clinical care and advocacy will help advance treatments and, ultimately, better long‑term prospects for all those affected by muscular dystrophy.
Whether newly diagnosed or managing long‑term challenges, UK muscular dystrophy charities are available to provide information, emotional support and practical guidance from teams who understand.
Book a personalised demonstration of the StandSure™ today by filling out our request form. Have any questions? Call us now, and our team will be happy to assist!



